



The Problem Everyone Pretends Doesn’t Exist
Chronic myeloid leukemia patients are told that tyrosine kinase inhibitors (TKIs) are miracle drugs. And for many, they are—until they’re not. What nobody mentions in the hopeful brochures is that 20-30% of CML patients develop resistance to front-line treatments like imatinib within five years.
The resistance isn’t random. It’s driven by specific mutations in the BCR-ABL1 fusion protein—particularly the T315I mutation, which acts like a molecular padlock that existing drugs simply cannot pick.
Enliven Therapeutics’ investigational drug ELVN-001 represents a fundamentally different approach to this lock-picking problem, and the early Phase 1 data suggests it might succeed where predecessors failed. But let me be clear about what “success” actually means in this context.
What Makes ELVN-001 Mechanistically Different
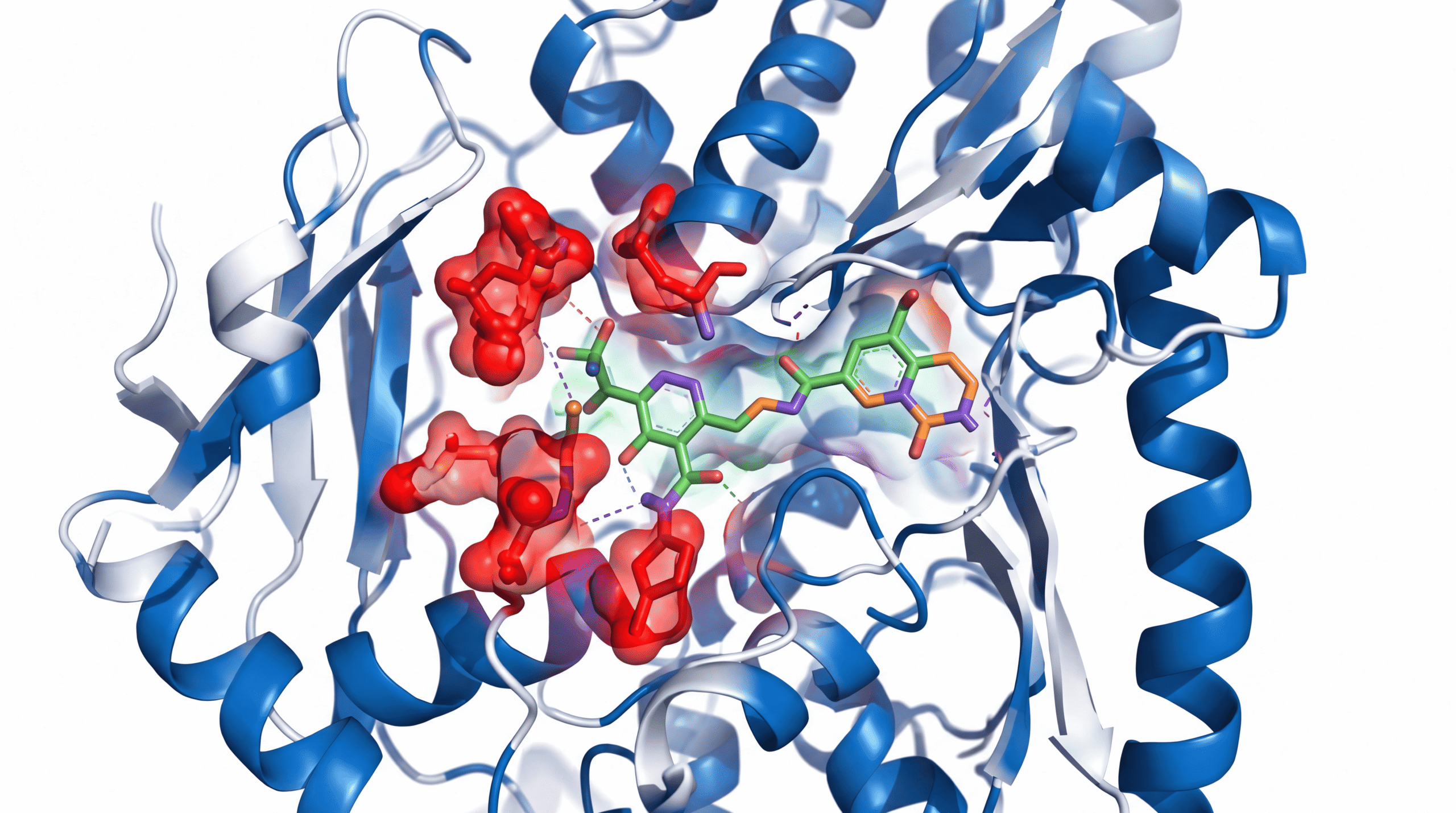
ELVN-001 is a next-generation BCR-ABL1 tyrosine kinase inhibitor, but that label undersells what it actually does. Unlike earlier generations of TKIs, this compound was specifically engineered to maintain binding affinity even when the BCR-ABL1 protein undergoes conformational changes caused by resistance mutations.
The T315I mutation—often called the “gatekeeper mutation”—replaces a threonine amino acid with isoleucine at position 315 of the ABL kinase domain. This single substitution sterically blocks most TKIs from accessing their binding site. It’s molecular armor.
ELVN-001 bypasses this by utilizing a different binding mode. According to preclinical crystallography studies, it maintains potency against T315I and compound mutations (like T315I plus additional mutations) that render patients essentially untreatable with current options.
The Early Clinical Data: What It Actually Shows
The Phase 1 ELVN-001-1001 trial enrolled heavily pre-treated CML patients—people who had failed a median of three prior TKI therapies. These aren’t ideal candidates. These are patients with limited options and aggressive disease biology.
In this population, preliminary data showed major cytogenetic responses in approximately 40% of evaluable patients with resistant mutations. Let me translate: their Philadelphia chromosome-positive cells (the hallmark of CML) decreased to less than 35% of bone marrow cells.
That might sound modest, but context matters. These patients had BCR-ABL1 mutations that made them resistant to ponatinib—previously considered the most potent available TKI. Achieving any response in this group represents a genuine biological breakthrough.
The drug also demonstrated a manageable safety profile, with thrombocytopenia (low platelets) and increased lipase being the most common Grade 3+ adverse events. Cardiovascular toxicity—a major limitation of ponatinib—appeared less frequent, though longer follow-up is needed.
What The Media Consistently Gets Wrong About Leukemia Drugs
The mainstream coverage of cancer drugs follows a predictable pattern: “Promising new drug shows hope for cancer patients.” This framing is both true and wildly misleading.
First mistake: conflating all leukemias. CML is a myeloproliferative neoplasm with a specific molecular driver (BCR-ABL1). Acute lymphoblastic leukemia, acute myeloid leukemia, and chronic lymphocytic leukemia are entirely different diseases with different biology, prognosis, and treatment paradigms. ELVN-001 is not relevant to most leukemias.
Second mistake: treating “response” and “cure” as synonyms. A major cytogenetic response means disease burden decreased substantially—not that it disappeared. Most CML patients on TKIs require indefinite treatment. Stopping therapy typically leads to molecular relapse in 50-60% of cases within two years, even after achieving deep molecular responses.
Third mistake: ignoring the denominator. When you read “40% response rate,” ask immediately: 40% of how many patients? In this Phase 1 trial, we’re talking about dozens of patients, not hundreds. The confidence intervals around that estimate are wide.
Fourth mistake: assuming approval equals accessibility. Even if ELVN-001 gains FDA approval based on confirmatory trials, the drug will likely cost $150,000-$200,000 annually. Insurance coverage battles will be substantial, particularly for a third- or fourth-line therapy.
The Biological Mechanism: Why Resistance Happens And What ELVN-001 Does About It
Understanding resistance requires understanding kinase biology. The BCR-ABL1 fusion protein is a constitutively active tyrosine kinase—meaning it’s stuck in the “on” position, continuously signaling cells to proliferate.
TKIs work by occupying the ATP-binding pocket of the kinase domain, preventing phosphorylation reactions that drive cellular proliferation. But the kinase domain is flexible. It exists in different conformational states: inactive, active, and intermediate forms.
First-generation TKIs like imatinib preferentially bind the inactive conformation. When mutations stabilize the active conformation or block drug access to the binding pocket, resistance emerges. This explains why the T315I mutation is so problematic—it physically prevents imatinib, dasatinib, and nilotinib from fitting into their binding site.
Second-generation TKIs like dasatinib and nilotinib address some of these mutations but remain vulnerable to T315I. Ponatinib, a third-generation TKI, was designed specifically to overcome T315I through a different binding approach. It largely succeeded but brought significant cardiovascular toxicity—likely related to off-target kinase inhibition of VEGFR and other vascular signaling pathways.
ELVN-001 represents a fourth-generation approach. According to biochemical studies, it maintains low nanomolar potency against native BCR-ABL1 and a comprehensive panel of resistance mutations, including compound mutations that combine T315I with secondary changes. Equally important, it demonstrates improved kinase selectivity—meaning less off-target inhibition of kinases involved in cardiovascular function.
What Success Actually Looks Like In Resistant CML
Let’s establish realistic expectations. “Success” for ELVN-001 does not mean cure. It means disease control in patients who have exhausted other options.
The primary endpoint in confirmatory trials will likely be major cytogenetic response by 12 months—getting Philadelphia chromosome-positive cells below 35%. A secondary endpoint will be complete cytogenetic response (0% Ph+ cells) and major molecular response (BCR-ABL1 transcript levels below 0.1% on the international scale).
If 50-60% of patients with resistant mutations achieve major cytogenetic response, and 30-40% achieve major molecular response, ELVN-001 would represent a substantial therapeutic advance. These patients would transition from having progressive, fatal disease to having controlled, chronic disease.
That matters enormously to individual patients. But it’s not the revolutionary transformation that media coverage often implies.
Long-term outcomes will depend on durability of response. Do patients maintain responses for years, or does resistance emerge again within 12-24 months? The Phase 1 data cannot yet answer this question. Median follow-up is measured in months, not years.
The Real Question: How Likely Is This Drug To Succeed?
Let’s assess probability honestly. ELVN-001 has several factors working in its favor.
First, clear unmet need. Patients with T315I-mutated CML have limited options after ponatinib failure. The FDA is likely to view favorably any drug that demonstrates meaningful activity in this population, particularly with manageable toxicity.
Second, rational design. This isn’t a drug that stumbled into activity against resistant mutations by accident. It was engineered specifically for this purpose, with structural biology guiding compound optimization. That doesn’t guarantee success, but it improves the odds.
Third, the mechanism is validated. We know that BCR-ABL1 inhibition controls CML. We’re not testing a novel biological hypothesis—we’re testing whether a better version of an established approach can overcome specific resistance mechanisms.
However, significant risks remain. Phase 1 data is inherently limited. Response rates often decrease as trials expand into larger, more heterogeneous patient populations in Phase 2 and 3 studies. The 40% response rate seen in highly selected Phase 1 patients might translate to 25-30% in a broader Phase 3 cohort.
Toxicity patterns also evolve with longer exposure and larger sample sizes. Rare but serious adverse events may only become apparent once hundreds of patients have been treated for extended periods. Cardiovascular events, in particular, may take 12-24 months to manifest clearly.
Realistically, I’d estimate a 60-70% probability that ELVN-001 achieves regulatory approval for resistant CML, assuming the Phase 2 data confirms Phase 1 findings. That’s higher than the typical oncology drug (roughly 30% of drugs entering Phase 2 ultimately gain approval), reflecting the strength of the preclinical rationale and early clinical data.
But approval probability and clinical impact are different questions. Even if approved, ELVN-001 will likely benefit 15,000-25,000 CML patients globally who have resistant disease—a meaningful number for those individuals and their families, but a small fraction of the broader cancer population.
What The Biology Tells Us About Future Resistance
Here’s what keeps me up at night as a clinician: resistance is not a problem you solve once. It’s an evolutionary process.
BCR-ABL1-positive cells are under constant selective pressure when exposed to TKI therapy. Any cell that acquires a mutation conferring survival advantage will expand clonally. ELVN-001 will exert different selective pressure than ponatinib, potentially selecting for different resistance mutations.
We’ve already seen this pattern with EGFR inhibitors in lung cancer. First-generation drugs selected for T790M resistance mutations. Third-generation drugs like osimertinib were designed to overcome T790M—and promptly selected for C797S mutations that confer resistance to osimertinib.
The solution isn’t a single perfect drug. It’s likely combination strategies—using multiple TKIs simultaneously to prevent emergence of resistant clones, similar to how we treat HIV with combination antiretroviral therapy. Or sequential therapy algorithms that anticipate specific resistance pathways.
Some of the most exciting research in CML now focuses on allosteric inhibitors—drugs that bind outside the ATP pocket and lock the kinase in an inactive state regardless of mutation status. If ELVN-001 could be combined with an allosteric inhibitor, we might achieve more durable disease control.
What You Should Actually Do If You Have Resistant CML
If you’re a CML patient who has developed resistance to your current TKI, here’s what you need to know.
First, get mutation testing. Not all resistance is mutation-driven—some is pharmacokinetic (inadequate drug levels due to absorption issues or drug interactions) or related to disease biology beyond BCR-ABL1. Knowing your specific mutation profile should guide treatment decisions.
Second, if you have T315I or compound mutations, discuss whether you’re a candidate for clinical trials of ELVN-001 or other investigational agents. Trials of ELVN-001 are actively enrolling at multiple academic medical centers. Your community oncologist may not be aware of these options—ask for referral to a CML specialist at an academic center.
Third, don’t wait until you’ve completely failed your current therapy to explore next steps. Patients with rising BCR-ABL1 transcripts despite adherence to therapy should start planning transitions early, while disease burden is still relatively low. Trying to rescue patients with blast crisis is far more challenging than managing chronic phase disease.
Fourth, consider allogeneic stem cell transplantation if you’re young and fit with a suitable donor. Transplant remains the only proven curative therapy for CML. In the TKI era, it’s reserved for advanced disease or multiple TKI failures—but for appropriate candidates, it offers a chance at genuine cure rather than indefinite drug dependence.
Finally, participate in systematic monitoring. Patients on any TKI should have BCR-ABL1 transcript levels measured by quantitative PCR every three months. Rising transcripts are the earliest signal of emerging resistance—often 6-12 months before frank hematologic relapse. Early detection allows earlier intervention.
The Bottom Line
ELVN-001 represents genuine progress in treating resistant CML, not because it’s a miracle cure, but because it addresses a specific molecular problem that existing drugs cannot solve. The early data is encouraging, the mechanism is rational, and the unmet need is real. But success in Phase 1 doesn’t guarantee success in Phase 3, and regulatory approval doesn’t equal accessibility or cure. If this drug ultimately reaches patients, it will extend lives and control disease—which is meaningful, even if it’s not revolutionary. The real transformation in cancer care doesn’t come from single breakthrough drugs; it comes from incremental improvements that, collectively, turn fatal diseases into manageable chronic conditions.


